Enzima

Modelo da enzima Purina nucleósido fosforilase (PNP) gerado por computador.
Enzimas são grupos de substâncias orgânicas de natureza normalmente proteica (existem também enzimas constituídas de RNA,[1] as ribozimas), com atividade intra ou extracelular que têm funções catalisadoras, catalisando reações químicas que, sem a sua presença, dificilmente aconteceriam. Isso é conseguido através do abaixamento da energia de ativação necessária para que se dê uma reação química, resultando no aumento da velocidade da reação e possibilitando o metabolismo dos seres vivos. A capacidade catalítica das enzimas torna-as adequadas para aplicações industriais, como na indústria farmacêutica ou na alimentar.
Em sistemas vivos, a maioria das reações bioquímicas dá-se em vias metabólicas, que são sequências de reações em que o produto de uma reação é utilizado como reagente na reação seguinte. Diferentes enzimas catalisam diferentes passos de vias metabólicas, agindo de forma concertada de modo a não interromper o fluxo nessas vias. Cada enzima pode sofrer regulação da sua actividade, aumentando-a, diminuindo-a ou mesmo interrompendo-a, de modo a modular o fluxo da via metabólica em que se insere. Sem as enzimas as reações químicas do organismo seriam muito lentas, elas proporcionam uma maior eficiência reduzindo a energia de ativação sem deslocar o equilíbrio exercendo assim seu poder catalítico.
Existem quatro tipos estruturais de enzimas: enzimas simples, enzimas alostéricas, enzimas complexas(holoenzima) e as isoenzimas.
O ramo da Bioquímica que trata do estudo das reações enzimáticas é a enzimologia. É comum ouvirmos falar sobre as enzimas, do quanto elas são importantes no nosso organismo e da sua função catalisadora no nosso sistema biológico. Porém, de fato, o que vem a ser uma enzima? Qual seu real papel e o que ela representa no nosso organismo?
Normalmente nosso organismo se depara com um sistema orgânico lento e pouco espontâneo, que depende exclusivamente do trabalho das chamadas enzimas para que possa ter um funcionamento metabólico melhor, de maneira mais regular e específica. Dizemos que essas enzimas funcionam como catalisadores celular, pois elas facilitam com que aconteçam certas reações internas de nosso corpo, dando uma melhor agilidade quando de certa forma as coisas costumam ocorrer mais devagar. Se não fossem as enzimas, diversas reações do nosso metabolismo aconteceriam de maneira exageradamente lenta, o que iria prejudicar e muito o nosso sistema. Elas agilizam as reações químicas das células aumentando a velocidade com que elas trabalham, assim, a sua atuação se torna mais eficaz, fazendo com que haja um melhor desempenho.
Índice
1 Atividade enzimática
2 Localização
3 História
4 Estruturas e mecanismos
4.1 Especificidade
4.1.1 Modelo chave-fechadura ( Não é mais utilizado )
4.1.2 Modelo do encaixe induzido
4.2 Mecanismos
4.2.1 Dinâmica e funções
5 Cofatores e coenzimas
5.1 Cofactores
5.2 Coenzimas
6 Termodinâmica
7 Cinética
8 Inibição
8.1 Usos de inactivadores
9 Funções biológicas
10 Controle da atividade
11 Envolvimento em doenças
12 Nomenclatura
12.1 Classes de enzimas
13 Aplicações
14 Referências
15 Ver também
Atividade enzimática |
As enzimas convertem uma substância, chamada de substrato, noutra denominada produto, e são extremamente específicas para a reação que catalisam. Isso significa que, em geral, uma enzima catalisa um e só um tipo de reacção química. Consequentemente, o tipo de enzimas encontradas numa célula determina o tipo de metabolismo que a célula efetua.
A velocidade da reação catalisada por uma enzima é aumentada devido ao abaixamento da energia de ativação necessária para converter o substrato no produto. O aceleramento da reação pode ser da ordem dos milhões de vezes: por exemplo, a enzima orotidina-5'-fosfato descarboxilase diminui o tempo da reação por ela catalisada de 78 milhões de anos para 25 milissegundos.[2]
Como são catalisadores, as enzimas não são consumidas na reação e não alteram o equilíbrio químico dela.
A atividade enzimática pode depender da presença de determinadas moléculas, genericamente chamadas cofatores. A natureza química dos cofatores é muito variável, podendo ser, por exemplo, um ou mais iões metálicos (como o ferro), ou uma molécula orgânica (como a vitamina B12). Estes cofatores podem participar ou não diretamente na reação enzimática.
Determinadas substâncias, podem inibir a atividade de algumas enzimas, diminuindo-a ou eliminando-a totalmente; são os chamados inibidores enzimáticos.
Pelo fato de serem proteínas com estrutura terciária ou quaternária, as enzimas são dotadas de dobramentos tridimensionais em suas cadeias polipeptídicas, o que lhes confere uma forma característica e exclusiva. Assim, diferentes enzimas têm diferentes formas e, portanto, diferentes papéis biológicos. Para que um enzima atue, é necessário que os substratos "se encaixem" na enzima. Esse "encaixe", porém, depende da forma, isto é, do "contorno" da enzima. Por isso, substratos que se "encaixam" em uma determinada enzima não se "encaixam" em outras diferentes, e a reação não ocorre; daí a especificidade das enzimas quanto aos substratos em que atuam. Uma vez ocorrido o "encaixe", forma-se o complexo enzima-substrato, que se assemelha ao sistema "chave-fechadura". O local da enzima onde o substrato se "encaixa" é denominado sítio ativo (ou centro ativo). No caso de substâncias que reagem entre si, sob a ação catalisadora das enzimas, a reação é facilitada, tornando-se mais rápida, pois a proximidade entre as moléculas "encaixadas" acelera o processo reativo; após a reação, a enzima desliga-se do substrato e permanece intacta.
Localização |
Podemos encontrar enzimas em quase todas as estruturas celulares e fluidos corporais. Algumas têm uma localização específica, de tal modo que podem servir para indicar que estamos em presença de um tal tecido, secreção ou fragmento celular se verificar a actividade de uma dada enzima.
História |

Eduard Buchner
Era já sabido, entre o final do século XVII e início do século XVIII, que secreções estomacais eram capazes de digerir a carne;[3] era também conhecida a conversão de amido a açúcares pela saliva e extractos vegetais. O mecanismo subjacente a estas transformações não era, no entanto, conhecido.[4]
As enzimas foram descobertas no século XIX, aparentemente por Pasteur, que concluiu que a fermentação do açúcar em álcool pela levedura é catalisada por fermentos. Ele postulou que esses fermentos (as enzimas) eram inseparáveis da estrutura das células vivas do levedo. Pasteur declarou que "a fermentação alcoólica é um acto correlacionado com a vida e organização das células do fermento, e não com a sua morte ou putrefacção".[5]
Em 1878, Wilhelm Kühne empregou pela primeira vez o termo "enzima" para descrever este fermento, usando a palavra grega ενζυμον, que significa "levedar". O termo passou a ser mais tarde usado apenas para as proteínas com capacidade catalítica, enquanto que o termo "fermento" se refere à atividade exercida por organismos vivos.
Em 1897, Eduard Buchner descobriu que os extratos de levedo podiam fermentar o açúcar até álcool e provou que as enzimas envolvidas na fermentação continuavam funcionando mesmo quando removidas das células vivas.[6] Esta descoberta valeu-lhe o prémio Nobel de Química em 1907.
Restava determinar qual a natureza das enzimas. Alguns afirmavam que as proteínas, associadas à actividade enzimática, apenas eram o suporte da verdadeira enzima, e, por si próprias, incapazes de catálise. Em 1926, James B. Sumner purificou e cristalizou a urease, mostrando tratar-se de uma proteína pura, e fez o mesmo, em 1937, para a catalase. A prova final foi feita por Northrop e Stanley em 1930, com o estudo de três enzimas digestivas, a pepsina, a tripsina e a quimotripsina, pelo que receberam o Prémio Nobel da Química em 1946.[7]
J.B.S. Haldane escreveu um tratado intitulado "Enzimas", onde continha a notável sugestão de que as interações por ligações fracas, entre a enzima e seu substrato, poderiam ser usadas para distorcer a molécula do substrato e catalisar a reação.
A cristalização de enzimas purificadas permitiu que as suas estruturas moleculares pudessem ser examinadas por cristalografia de raios X, o que aconteceu primeiro com a lisozima, uma enzima que ocorre na saliva, lágrimas e na clara de ovo e destrói a parede celular de bactérias, em 1965.[8] Começaram assim a bioquímica e biologia estruturais, que se esforçam por compreender o funcionamento dos enzimas a nível atómico.
A tentativa de compreender o mecanismo da catálise enzimática a nível quântico tem sido facilitada recentemente pelo aumento de capacidade aritmética dos computadores.
Estruturas e mecanismos |

Diagrama PDB 1MOO mostrando a Anidrase carbónica II. A esfera a cinzento é um ião de zinco que funciona como cofactor no sítio activo.
As enzimas são proteínas, e podem ter um tamanho desde 62 resíduos de aminoácidos, como é o caso do monómero da enzima 4-oxalocrotonato tautomerase,[9] até um tamanho de 2.500 resíduos, como é o caso da sintase de ácidos gordos.[10] A atividade das enzimas são determinadas pela sua estrutura quaternária.[11] A maioria das enzimas são maiores do que o substrato sobre o qual atuam, e só uma pequena porção da enzima (cerca de 3-4 aminoácidos) está envolvida na catálise.[12] A região que contém estes resíduos catalíticos, que se liga-se ao substrato e que desempenha a reação, é denominada de sítio activo. As enzimas também podem ter sítios onde se ligam cofactores, que são necessários às reações catalíticas. Algumas enzimas também podem ter sítios de ligações para pequenas moléculas, que são produtos ou substratos, diretos ou indirectos, da reação catalisada. Estas ligações servem para aumentar ou diminuir a atividade da enzima, providenciando um meio de regulação por feedback.
Tal como todas as proteínas, as enzimas são formadas por longas cadeias lineares de aminoácidos que sofrem um enovelamento que tem como resultado um produto com estrutura tridimensional. Cada sequência única de aminoácidos produz também uma estrutura tridimensional única que tem propriedade específicas. Cadeias individuais de proteínas podem por vezes agrupar-se para formar um complexo proteico. A maioria das enzimas pode sofrer desnaturação, isto é, a sua estrutura pode sofrer desagregação e inativação pelo aumento de temperatura, o que provoca alterações na conformação tridimensional da proteína. Dependendo da enzima, a desnaturação pode ter efeitos reversíveis ou irreversíveis.
Como toda proteína, as enzimas também são sensíveis a variações de pH. O pH pode alterar fatores como a ligação do substrato à enzima, a atividade catalítica da enzima, a ionização do substrato e a variação da estrutura da proteína. [13][14]
As cadeias laterais dos aminoácidos no sítio ativo das enzimas podem agir como ácidos fracos ou bases com funções críticas para a manutenção de um estado ionizado necessário para a atividade enzimática. A faixa de pH na qual a enzima sofre alterações em sua atividade pode indicar inclusive o tipo de resíduo de aminoácido envolvido na catálise. [13][14]
Portanto, o pH da solução influencia a atividade enzimática, e, consequentemente, a velocidade das reações: se o pH está em sua faixa ótima para a ação de determinada enzima, a velocidade da catálise será mais rápida. É importante ressaltar que a velocidade e a eficiência da catálise enzimática são influenciadas por vários outros fatores, como temperatura, regulação da enzima, presença ou ausência de cofatores, etc. [13][14]

Relação entre os valores de pH e a velocidade da reação e os valores dos pH ótimos para algumas enzimas.
Especificidade |
As enzimas possuem normalmente uma alta especificidade em relação às reações que catalisam e aos substratos que estão envolvidos nessas reações. A forma complementar, carga e características hidrofílicas/hidrofóbicas, são responsáveis por esta especificidade. As enzimas exibem também elevados níveis de estereoespecificidade, regioselectividade e quimioselectividade.[15]
Algumas das enzimas que apresentam maior especificidade e precisão, estão envolvidas na cópia e expressão do genoma. Estas enzimas possuem mecanismos de proof-reading (revisão). Um destes casos é a DNA polimerase, que catalisa uma reacção num primeiro passo, para de seguida confirmar, num segundo passo, se o produto é o correcto.[16] Este processo em duas etapas resulta em médias de taxa de erro muito diminutas, na ordem de uma para cem milhões de reacções, no caso de polimerases de mamíferos.[17] Mecanismos de revisão similares também podem ser encontrados na RNA polimerase,[18] na aminoacil-tRNA sintetases[19] e em ribossomas.[20]
Algumas enzimas que produzem metabolitos secundários são descritos como promíscuos, visto que podem actuar num largo espectro de diferentes substratos. Tem sido sugerido que este tipo de especificidade alargada é importante nos processos de evolução de novas vias de biossíntese.[21]
Modelo chave-fechadura ( Não é mais utilizado ) |
As enzimas exibem uma elevada especificidade, e foi sugerido por Emil Fischer, em 1894, que esse facto era devido a que tanto as enzimas como os substratos apresentam formas geométricas complementares, fazendo com que encaixem de maneira precisa umas nos outros.[22] Este processo é muitas vezes referido como modelo chave-fechadura. No entanto, apesar deste modelo explicar a especificidade das enzimas, falha em explicar a estabilização dos estados de transição que as enzimas exibem.
Modelo do encaixe induzido |

Diagramas que mostram a actividade enzimática através do modelo do encaixe induzido.
Em 1958, Daniel Koshland sugeriu uma modificação ao modelo de chave-fechadura: uma vez que as enzimas exibem estruturas flexíveis, o sítios activo altera a sua forma de maneira continuada através de interacções com o substrato, enquanto esse mesmo substrato vai interagindo com a enzima.[23] Como resultado, o substrato não se liga simplesmente a um sítio activo que é rígido. As cadeias laterais dos aminoácidos que formam o sítios activo sofrem um reorientação de maneira a que as suas posições potenciem a acção catalítica da enzima. Em alguns casos, como nas glicosidases, a molécula de substrato também sofre alterações de conformação à medida que vai se aproximando do sítio activo.[24] O sítio activo continua a sofrer modificações até que o substrato esteja completamente ligado e é neste momento em que a conformação final e a carga são determinadas.[25]
Mecanismos |
As enzimas actuam de diversas formas, todas elas baixando o valor de ΔG‡:[26]
- Baixando a energia de activação, através da criação de um ambiente no qual o estado de transição é estabilizado (por exemplo, distorcendo a forma da molécula do substrato - a enzima distorce o substrato, gastando energia neste passo, de modo a baixar a energia do estado de transição da reacção catalisada, resultando numa diminuição global da energia requerida para completar a reacção).
- Providenciando uma via alternativa (por exemplo, reagindo com o substrato formando um complexo enzima-substrato, de existência impossível sem a presença da enzima).
- Reduzindo a variação da entropia da reacção ao orientar os substratos de forma correcta para facilitar a reacção. Na ausência de enzima, as moléculas colidem em todas as direcções possíveis de forma aleatória, um processo menos eficiente que na presença da enzima. Ao considerar-se ΔH‡ isoladamente, este aspecto é negligenciado.
O mecanismo de regulação da atividade enzimática pode ser lento, quando a regulação apresenta aumento ou diminuição de síntese de enzimas, ou rápido que é por meio molecular, ou seja, por ligações de moléculas na enzima ou no substrato, podendo ser também por ligações covalentes “fosforilação”.[27]
Dinâmica e funções |
Investigações recentes providenciaram novos conhecimentos sobre a ligação entre a dinâmica interna de uma enzima e o seu mecanismo de catálise.[28][29][30] A dinâmica interna de uma enzima é descrita como o movimento de partes internas (como aminoácidos individuais, grupos de aminoácidos, um laço da cadeia, uma hélice alfa, folhas beta vizinhas ou até domínios proteicos inteiros) destas biomoléculas, que podem ocorrer a diversas escalas de tempo, desde femtossegundos a segundos. Redes de resíduos de aminoácidos de uma estrutura podem contribuir para a catálise através de movimentos dinâmicos.[31][32][33][34] Os movimentos em proteínas são importantes para diversas enzimas mas o tipo de reacção que elas catalisam determina que tipos de movimento são mais importantes: pequenas e rápidas vibrações ou lentas e significativas alterações conformacionais. Estes estudos têm consequências na compreensão dos efeitos alostéricos, na produção industrial de enzimas e no desinadas moléculas. A modulação da actividade da enzima pode ser directa, quando a molécula se liga directamente a um local de ligação na enzima, ou indirecta, quando a molécula se liga a outras proteínas ou subunidades que interagem com a enzima alostérica, influenciando então a sua actividade.
As enzimas alostéricas são também, em geral, maiores e mais complexas que as enzimas simples. A maioria tem duas ou mais cadeias polipeptídicas ou subunidades.
A maioria das enzimas, em cada sistema, segue os padrões cinéticos já descritos. Em cada sistema enzimático, entretanto, há pelo menos uma enzima que determina a velocidade de toda a sequência, porque ela catalisa a reação mais lenta ou a reação limitante da velocidade. Estas enzimas reguladoras têm sua atividade catalítica aumentada ou diminuída em resposta a determinados sinais. Pela ação destas enzimas reguladoras a velocidade de cada sequência metabólica é constantemente ajustadas as mudanças que ocorrem nas necessidades de energia e de biomoléculas requeridas no crescimento da célula e no seu auto-reparo.[35]
A enzima alostérica possui ,além do sítio ativo específico, um sítio de ligação de moléculas, denominado sítio alostérico. Nesse sítio pode se ligar os produtos da reação para realizar um “feedback” negativo da reação e regulá-la melhor, podendo se ligar também efetores (inibidores) que aumentam ou diminuem a afinidade da enzima e substrato. O substrato pode se ligar ao sítio alostérico, denominado fenômeno de cooperação, o que dá característica ao gráfico das enzimas alostéricas.
Existem também enzimas que atuam com o mesmo substrato realizando a mesma reação, porém com algumas diferenças. Essas possuem sítio ativo específico para o mesmo substrato, entretanto apresentam regulações alostéricas distintas, são denominadas isoenzimas.[36]
Cofatores e coenzimas |
Cofactores |
Algumas enzimas não necessitam de componentes adicionais para mostrarem uma actividade completa. No entanto, outras requerem ligação a moléculas não proteícas para que possam exercer a sua actividade. Os cofactores podem ser inorgânicos (iões metálicos e complexos ferro-enxofre) ou compostos orgânicos (flavina ou heme). Os cofactores orgânicos (coenzimas) são normalmente grupos prostéticos, que estão intimamente ligados às enzimas a que eles prestam assistência. Os cofactores que possuem ligação forte às enzimas são distintos de outras coenzimas, tal como a NADH, visto que não são libertados do sítio activo durante a reacção catalisada.
Um exemplo de uma enzima que contém um cofactor é a anidrase carbónica, que é mostrada em figura acima com um ião de zinco como cofactor.[37] Estas moléculas que possuem uma ligação estreita com as enzimas são normalmente encontradas no sítio activo e estão envolvidas na reacção catalítica. Por exemplo, a flavina e grupos heme estão muitas vezes envolvidos em reações de oxidação-redução.
Enzimas que requerem um cofactor mas que não se ligam a estes, são denominadas apoenzimas. Uma apoenzima juntamente com o(s) seu(s) cofactor(es) são denominadas holoenzimas. As enzimas complexas, ou holoenzimas, possuem além da parte protéica (apoenzima), um cofator, podendo ser um íon ou uma coenzima, que é uma molécula orgânica, vitamina ativada transformando-se em coenzima.
A maioria dos cofactores não se ligam por covalência a uma enzima, mas estabelecem ligações fortes. No entanto, os grupos prostéticos orgânicos podem ligar-se de maneira covalente. Um exemplo disso é a ligação da tiamina pirofosfato à enzima piruvato desidrogenase.
O bom funcionamento do organismo se deve ao fato de que alguns cofatores terem a habilidade de se separarem de suas respectivas apoenzimas e operar funções específicas. O exemplo mais conhecido para este fato é o NAD e o FAD, ambas coenzimas de holoenzimas desidrogenases que, ao capturarem hidrogênio, separam-se da apoenzima para terem importante função no ciclo de Krebs.
Coenzimas |

Modelo tridimensional da coenzima NADH.
As coenzimas são pequenas moléculas que transportam grupos químicos de uma enzima para outra.[38] Alguns destes compostos químicos, como a riboflavina, a tiamina e o ácido fólico, são vitaminas, compostos que não são sintetizados no organismo e que têm de ser assimilados através da dieta. Os grupos químicos transportados incluem o ião hidreto (H-) transportado pelo NAD ou NADP+, o grupo acetilo transportado pela coenzima A, os grupos formilo, metenilo e metilo transportados pelo ácido fólico e o grupo metilo transportado pela S-adenosilmetionina.
Dado que as coenzimas são modificadas quimicamente como consequência da acção enzimática, é útil considerá-las como sendo uma classe especial de substratos, ou substratos secundários, que são comuns em muitos tipos de enzimas diferentes. Por exemplo, sabe-se que existem cerca de 700 enzimas que utilizam a coenzima NADH.[39]
As coenzimas sofrem regeneração e as suas concetrações são mantidas a um nível estável dentro da célula. Por exemplo, o NADPH é regenerado através da via das pentoses-fosfato e a S-adenosilmetionina é regenerada pela metionina adenosiltransferase.
Termodinâmica |

Diagrama de uma reacção catalítica, mostrando o nível de energia em cada etapa da reacção. Normalmente, o substrato necessita de uma quantidade elevada de energia para conseguir chegar ao estado de transição, decaindo depois até a um produto final. A enzima estabiliza o estado de transição, reduzindo o valor de energia necessário para que se formem os produtos. A linha a vermelho (cheio) representa a reacção na ausência de catalisador; a azul (tracejado), na presença de enzima. S: nível de energia do(s) substrato(s); P: nível de energia do(s) produto(s); G: energia de Gibbs; R: coordenada de reacção (sentidos de progressão de reacção); ΔG'º: energia livre padrão bioquímica; ΔG‡: energia de activação (S-P: do(s) substrato(s); P-S: do(s) produto(s)).
Tal com acontece com todos os compostos catalisadores, as enzimas não alteram a posição do equilíbrio químico da reacção. Normalmente, na presença de uma enzima, a reacção desenvolve-se na mesma direção que se seria tomada se ela não estivesse presente, mas de maneira mais rápida. no entanto, Na ausência da enzima, as reações poderão dar origem a diferentes produtos, porque nessas condições esses produtos são formados de maneira mais rápida.
Para além disso, as enzimas podem executar duas ou mais reacções, de tal maneira que a actuação numa reacção termodinamicamente favorável possa facilitar a actuação numa reacção menos favorável. Por exemplo, a energia disponibilizada pela hidrólise do ATP é normalmente utilizada para executar outras reacções.
As enzimas catalisam as reacções em ambos os sentidos. Elas não alteram o equilíbrio em si, mas a velocidade em que este é alcançado. Por exemplo, a anidrase carbónica catalisa a sua reacção nas duas direcções, dependendo da concentração dos seus reagentes.
CO2+H2OAnidrase carbonica→H2CO3{displaystyle mathrm {CO_{2}+H_{2}O{}^{mathrm {quad Anidrase carbonica} }!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!{overrightarrow {qquad qquad qquad qquad }}H_{2}CO_{3}} }(em tecidos; alta concentração de CO2)
H2CO3Anidrase carbonica→CO2+H2O{displaystyle mathrm {H_{2}CO_{3}{}^{mathrm {quad Anidrase carbonica} }!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!{overrightarrow {qquad qquad qquad qquad }}CO_{2}+H_{2}O} }(nos pulmões; baixa concentração de CO2)
Se o equilíbrio estiver substancialmente deslocado para uma das direcções, isto é, se for uma reacção muito exergónica, a reacção é efectivamente irreversível. Nestas condições, a enzima só catalisará a reacção na direcção termodinamicamente permitida.
Cinética |

Mecanismo de reacção de uma enzima com substrato único. A enzima (E) liga o substrato (S) e liberta o produto (P).
A cinética enzimática é o estudo do mecanismo pelo qual as enzimas ligam substratos e os transformam em produtos. São utilizados dados sobre a velocidade de reacção de enzimas através de ensaios enzimáticos.
Em 1902, Victor Henri[40] propôs uma teoria quantitativa de cinética enzimática, mas os seus dados experimentais tinham pouca utilidade porque não era então considerada a importância da concentração do ião H+. Depois de Peter Lauritz Sørensen definir a escala logarítmica de pH e introduzir o conceito de solução tampão em 1909,[41] o químico alemão Leonor Michaelis e a sua pós-doc canadiana Maud Leonora Menten repetiram as experiências de Henri e confirmaram a sua equação, sendo esta cinética conhecida como cinética de Henri-Michaelis-Menten (muitas vezes simplificado para cinética de Michaelis-Menten).[42] Este trabalho foi desenvolvido por G. E. Briggs e J. B. S. Haldane, que derivaram equações cinéticas usadas ainda hoje em dia.[43]
Henri contribuiu significativamente para este campo ao teorizar as reacções enzimáticas ocorrendo em dois passos. No primeiro, o substrato liga-se de forma reversível à enzima, formando o complexo enzima-substrato. Este complexo é por vezes designado complexo de Michaelis. A enzima catalisa então o passo químico da reacção e liberta o produto.
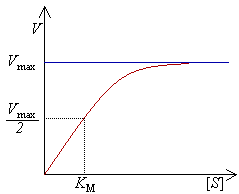
Curva de saturação numa reacção enzimática, mostrando a relação entre a concentração de substrato ([S]) e a velocidade (V).
As enzimas podem catalisar até vários milhões de reacções por segundo. Por exemplo, a reacção catalisada pela orotidina 5'-fosfato descarboxilase tem uma semivida de 78 milhões de anos na ausência de enzima, diminuindo para apenas 25 milissegundos na presença desta.[44] As velocidades de reacção dependem das condições em que as enzimas se encontram e da concentração de substrato. Condições de alta temperatura, pH extremos ou altas concentrações salinas desestabilizam a estrutura da proteína, desnaturando-a. Por outro lado, em geral, um aumento na concentração de substrato tende a aumentar a actividade enzimática.
Experimentalmete, encontra-se a velocidade máxima de reacção aumentando progressivamente a concentração de substrato, até se verificar uma velocidade constante de formação de produto. Tal é visível na curva de saturação do gráfico à direita. A saturação acontece porque, à medida que é aumentada a concentração de substrato, aumenta também a quantidade de enzima presente sob a forma de complexo enzima-substrato (ES). À velocidade máxima, Vmax, todos os centros activos estão ocupados (saturados) com substrato, ou seja, não existe enzima livre para ligar mais substrato e a concentração de complexo ES é igual à concentração de enzima.
A actividade de uma enzima é geralmente descrita através de Vmax e também da constante de Michaelis-Menten, KM, que representa a concentração de substrato à qual se detecta uma velocidade de reacção igual a metade de Vmax. Cada enzima possui um KM característico para um dado substrato; este parâmetro é muitas vezes usado para demonstrar a força de ligação de um substrato à enzima. É também usada outra constante cinética, kcat, para descrever o comportamento de uma enzima, representando o número de moléculas de substrato que podem ser catalisadas por centro activo por segundo.
A eficiência catalítica de uma enzima pode ser expressa através da constante de especificidade, igual à razão kcat/Km. A constante de especificidade relaciona-se tanto com a afinidade da ligação do substrato à enzima como com a capacidade catalítica desta, pelo que se torna útil para a comparação entre diferentes enzimas, ou a mesma enzima com diferentes substratos. Existe um valor máximo teórico para esta constante, cerca de 108 to 109 M−1 s−1, denominado limite de difusão. Considerando-se que cada colisão entre enzima e substrato resulta em catálise, a velocidade global de formação do produto não será limitada pela velocidade de reacção da enzima mas sim pela velocidade de difusão das moléculas em solução. Enzimas que possuam uma constante de especificidade perto deste valor são designadas enzimas cataliticamente (ou cineticamente) perfeitas; alguns exemplos incluem as enzimas triose fosfato isomerase, anidrase carbónica, acetilcolinesterase, catalase, fumarase, ß-lactamase e superóxido dismutase.
A cinética de Michaelis-Menten baseia-se na lei da acção das massas, que é derivada das assunções sobre difusão livre e sobre colisões aleatórias com base termodinâmica. No entanto, muitos dos processos bioquímicos ou celulares não se comportam da forma prevista por estes modelos devido à alta concentração de substâncias no meio celular, separação de fases entre enzima, substrato e produto e restrição do movimento molecular a uma ou duas dimensões.[45] Nestas situações, é aplicável um modelo fractal da cinética de Michaelis-Menten.[46][47][48][49]
Algumas enzimas apresentam cinética mais veloz que as velocidades de difusão, no que aparenta ser uma impossibilidade. Diversos mecanismos foram usados como razão para explicar este fenómeno. Uma explicação é a de algumas enzimas acelerarem a sua catálise ao captar e orientar o seu substrato usando dipolos eléctricos. Outros modelos usam um mecanismo quântico-mecânico de tunneling, em que um protão ou electrão podem atravessar barreiras de activação, embora o modelo de tunneling de protões seja controverso,[50][51] tendo sido no entanto observado na triptamina.[52] Estes modelos sugerem que a catálise enzimática seja possivelmente melhor descrita em termos de "atravessar um barreira" em detrimento do modelo tradicional de "passar por cima" de uma barreira energética diminuída.
Inibição |

Exemplo de uma reacção normal (a) e de uma inibição reversível competitiva (b), em que o substrato e o inibidor competem pela enzima. Numa reacção normal, o Substrato liga-se ao centro Activo da Enzima (1), provocando a libertação de produtos por parte da enzima (2). Quanto à inibição, o Inibidor liga-se à enzima (3), não permitindo ao substrato ter acesso à mesma (4), logo não havendo reacção e não sendo produzidos quaisquer produtos.
As taxas de reacção enzimáticas podem ser diminuídas por acção de vários tipos de inibidores enzimáticos.
- Inibição competitiva
Na inibição competitiva, o inibidor e o substrato competem reversivelmente ao mesmo sítio enzimático. Os inibidores são muitas vezes semelhantes ao substrato da enzima. Por exemplo, o metotrexato é um inibidor competitivo da enzima dihidrofolato redutase, que catalisa a redução do dihidrofolato em tetrahidrofolato. A similitude entre as estruturas do ácido fólico e desta droga são mostradas na figura adjacente. Nota-se que o local de ligação do inibidor não é necessariamente o mesmo que o local de ligação do inibidor, se a ligação de este último mudar a conformação da enzima com vista a impedir a ligação do substrato, e vice versa. Na inibição competitiva, a velocidade máxima da reação não é alterada, mas níveis mais altos de concentração do substrato são requeridos para que se atinja uma determinada velocidade, com o aumento do KM. Outro exemplo de inibidor competitivo são as estatinas, essas substâncias anti-hiperlipidêmicas inibem competitivamente a primeira reação na síntese do colesterol, inibindo a hidroximetilglutaril-CoA-redutase (HMG-CoA-redutase).[27]
- Inibição acompetitiva ou incompetitiva
Na inibição acompetitiva, o inibidor não se pode ligar à enzima no estado livre, mas somente ao complexo E-S. O complexo E-I-S assim formado, é enzimaticamente inactivo. Este tipo de inibição é raro, mas pode ocorrer em enzima multiméricas.
- Inibição não-competitiva
Os inibidores não-competitivos podem ligar-se à enzima ao mesmo tempo que o substrato, ou seja, nunca se ligam ao sítio ativo. Quer o complexo E-I, quer o complexo E-I-S, são enzimaticamente inactivos. O KM aparente não é alterado, a ligação do inibidor não interfere no sítio ativo da enzima, mas o Vmax muda neste caso, pois o inibidor não pode ser desligado da enzima por aumento da concentração de substrato (em contraste com o que acontece na inibição competitiva).
- Inibição mista
Este tipo de inibição assemelha-se à inibição não-competitiva. Neste caso, o complexo E-I-S possui actividade enzimática residual.
Em muitos organismos, os inibidores podem agir como parte do mecanismo de retroalimentação. Se uma enzima produz um determinada substância em demasia, essa substância poderá agir como inibidor da enzima, no início da via que a produz, causando a redução ou paragem da produção da substância quando esta se acumula. Esta é um forma de retroalimentação negativa. Enzimas que estão sujeitas a esta forma de regulação são muitas vezes multiméricas e possuem sítios de ligação alostéricos para substâncias reguladoras. Os seus gráficos substrato/velocidade não têm a forma hiperbólica mas sim forma sigmóide (curva em forma de S)
O ácido fólico (à esquerda) e a droga anticancerígena metotrexato são semelhantes na sua estrutura. Como resultado, o metotrexato é um inibidor competitivo de muitas das enzimas que utilizam os folatos.
Os inibidores irreversíveis reagem com a enzima e formam ligações covalentes com a cadeia polipeptídica. Este tipo de inactivação é irreversível. Um exemplo de inibidor irreversível é a eflornitina, uma substância utilizada no tratamento da doença do sono.[53] A penicilina e a aspirina também actuam deste modo. Nestes casos, o composto liga-se ao centro activo e a enzima converte-o a uma forma activada que reage irreversivelmente com um ou mais resíduos de aminoácidos.
Usos de inactivadores |
Os inactivadores são muitas vezes usados como medicamentos, mas também poderão actuar como venenos. No entanto, a diferença entre um medicamento e um veneno é normalmente uma questão de dosagem, visto que a maior parte dos medicamentos e drogas são tóxicas a partir de certo nível. Como Paracelso disse: "Todas as coisas são um veneno e nada existe sem veneno, apenas a dosagem é razão para que uma coisa não seja um veneno"[54] Igualmente, os antibióticos e outras drogas antiinfecciosas são apenas venenos que matam o agente patogénicos e não o hospedeiro deste.
Um exemplo de um inactivador que é usado como medicamento é a aspirina, que inibe a ciclooxigenase 1 e a ciclooxigenase 2, que são enzimas que produzem um mensageiro que age em casos de inflamação, neste caso uma prostaglandina, suprimindo assim a dor e a inflamação. O cianureto é um inactivador enzimático irreversível, que se combina com cobre e zinco no sítio activo da enzima citocromo c oxidase, bloqueando a respiração celular.[55]
Funções biológicas |

A acção da luciferase induz bioluminiscência nos pirilampos
As enzimas exercem uma grande variedade de funções nos organismos vivos. São indispensáveis para a transdução de sinais, na regulação celular, muitas vezes por acção de cinases e fosfatases.[56] Através da sua acção podem gerar movimento, como no caso da miosina que hidroliza ATP, gerando contracções musculares. Também movimentam carga através da célula, através da acção do citoesqueleto.[57] Algumas enzimas são ATPases (funcionam como bombas iónicas), que se localizam na membrana celular, estando envolvidas do processo de transporte activo. Algumas funções mais exóticas são operadas por enzimas, como é o caso da luciferase que gera luz nos pirilampos.[58] Os vírus podem conter enzimas que auxiliam na infecção de células (HIV-integrase e transcriptase reversa) ou na libertação celular de vírus (neuraminidase no vírus influenza).
Uma importante função das enzimas tem lugar no sistema digestivo dos animais. Enzimas como as amilases e proteases, quebram grandes moléculas como o amido e proteínas, respectivamente, em moléculas de menores dimensões, de maneira a que estas possam ser absorvidas no intestino. O amido não é absorvível no intestino, mas as enzimas hidrolizam as cadeias de amido em moléculas menores tais como a maltose e a glucose, podendo desta maneira ser absorvidas. Diferentes enzimas actuam sobre diferentes tipos de alimento. Nos ruminantes, que possuem uma dieta herbívora, bactérias no sistema digestivo produzem uma enzima denominada celulase que quebra as paredes celulares das células vegetais.
As enzimas podem trabalhar em conjunto, seguindo uma ordem de actuação específica. Desta maneira podem formar vias metabólicas. Nestas vias, uma enzima processa o produto da acção de outra enzima como o seu substrato. Após a reacção catalítica, o produto é depois entregue a outra enzima. Por vezes, mais do que uma enzima pode catalisar a mesma reacção, em paralelo. Isto permite uma regulação mais complexa:
As enzimas determinam que passos é que ocorrem nessa vias metabólicas. Sem a presença de enzimas, o metabolismo não progride através dos mesmo passos, nem é suficientemente rápido para que sirva as necessidades da célula. De facto, uma via metabólica tão importante como a glicólise não poderia existir sem a presença de enzimas. A glucose, por exemplo, pode reagir directamente com o ATP para dar origem a um produto fosforilado em um ou mais carbonos. Na ausência de enzimas, este processo é tão lento que se torna insignificante. No entanto, se a enzima hexocinase for adicionada, estas reacção lentas continuam a ser efectuadas, mas a fosforilação do carbono número 6 ocorre de maneira tão rápida que, se a mistura for testada pouco tempo depois, a glicose-6-fosfato o único produto significativo. Consequentemente, pode-se dizer que a rede de vias metabólicas existentes dentro de cada célula depende do conjunto de enzimas funcionais que estão presentes.
Controle da atividade |
A atividade enzimática na célula é controlada de cinco principais modos:
- A produção da enzima (transcrição e tradução dos genes que codificam a enzima) pode ser aumentada ou diminuída pela célula em resposta a mudanças no ambiente celular. Esta forma de regulação genética é designada como indução ou inibição da expressão enzimática. Por exemplo, as bactérias podem desenvolver resistência a antibióticos como a penicilina porque são induzidas enzimas (β-lactamases) que hidrolisam o anel beta-lactâmico presente na estrutura do antibiótico e essencial para a sua actividade biológica. Outro exemplo é a importância das enzimas hepáticas citocromo P450 oxidases, envolvidas no metabolismo de desintoxicação de xenobióticos.
- As enzimas podem encontrar-se compartimentadas, com difentes vias metabólicas (ou partes de vias metabólicas) ocorrendo em diferentes compartimentos celulares. Por exemplo, os ácidos gordos são sintetizados por um conjunto de enzimas no citoplasma, no retículo endoplasmático e no complexo de Golgi e usados por um conjunto diferente de enzimas como fonte de energia na mitocôndria, através da β-oxidação.[59]
- As enzimas podem ser reguladas por inibidores e activadores. Por exemplo, os produtos finais de uma dada via metabólica são frequentemente inibidores das enzimas que catalisam os primeiros passos da via (normalmente o primeiro passo factualmente irreversível), regulando assim a quantidade de produto final produzido pela via. Este tipo de regulação é denominado mecanismo de feedback (ou autoalimentação) negativo porque a quantidade de produto final é regulada pela sua própria concentração. Na prática, o feedback negativo ajusta a velocidade de síntese de metabolitos intermediários da via de acordo com a necessidade da célula. Este mecanismo ajuda na distribuição económica e eficiente de compostos, evitando a produção excessiva de produtos finais. Tal como outros mecanismos homeostáticos, o controlo da actividade enzimática ajuda na manutenção de um ambiente interno estável em organismos vivos.
- As enzimas podem ser reguladas através da sua modificação pós-traducional. Este tipo de modificação inclui a fosforilação, a miristoilação e a glicosilação. Por exemplo, a fosforilação de diversas enzimas, como a glicogénio sintase, em resposta a um sinal da insulina, ajuda no controlo da síntese ou degradação do glicogénio e permite a resposta celular a variações da glicemia.[60] Outro exemplo é a quebra da cadeia polipeptídica da quimotripsina, uma protease digestiva que é produzida numa forma inactiva (quimotripsinogénio) no pâncreas e transportada então para o estômago, onde é activada. Este mecanismo evita a digestão de tecidos pancreáticos (ou outros) pela quimotripsina antes de entrar no tracto digestivo. Este tipo de precursor inactivo de uma enzima é denominado zimógeno.
- Algumas enzimas tornam-se activas quando são colocadas num ambiente diferente (por exemplo, de um ambiente citoplasmático redutor para um ambiente periplasmático oxidativo). Um exemplo encontra-se na hemaglutinina dos vírus Influenza (vírus da gripe), que sofre uma mudança conformacional quando encontra o ambiente acídico de vesículas da célula hospedeira, provocando a sua activação.[61]
Envolvimento em doenças |

Fenilalanina hidroxilase. [http://www.rcsb.org/pdb/explore.do?structureId=1KW0 PDB 1KW0
]
Uma vez que controlo da actividade enzimática é essencial para a homeostase, qualquer alteração em genes que codifiquem enzimas (mutações ou delecções) poderá ter como efeito o aparecimento de doenças genéticas. A importância das enzimas é demonstrada pelo facto de que uma doença letal pode ser causada pelo mau funcionamento de um único tipo de enzima entre as milhares que estão presentes no corpo humano.
Um exemplo disso é que o acontece com o tipo mais comum de fenilcetonúria. Uma mutação num único aminoácido da enzima fenilalanina hidroxilase, que catalisa o primeiro passo na degradação da fenilalanina, resulta na acumulação da fenilalanina e dos produtos associados. Isto pode causar atraso mental se a doença não for tratada.[62]
Outro exemplo acontece quando mutações em genes que codificam enzimas envolvidas no reparo de DNA causam síndromas de cancro hereditário, tal com a xerodermia pigmentosa. Defeitos nestas enzimas podem causar cancro, visto que o corpo fica com uma habilidade menor para reparar mutações no genoma. Isto causa uma lenta acumulação de mutações e resulta no desenvolvimento de muitos tipos de cancro no paciente.
Nomenclatura |
A determinação do nome das enzimas é normatizada por um comitê especializado, o Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB).
Classes de enzimas |
| Classe 1 | Oxirredutases | Catalisam reacções de oxirredução, transferindo eletrons, hidretos (H-) ou protons (H+). |
| Classe 2 | Transferases | Transferem grupos químicos entre moléculas. |
| Classe 3 | Hidrolases | Utilizam a água como receptor de grupos funcionais de outras moléculas. |
| Classe 4 | Liases | Formam ou destroem ligações duplas, respectivamente retirando ou adicionando grupos funcionais. |
| Classe 5 | Isomerases | Transformam uma molécula num isómero. |
| Classe 6 | Ligases | Formam ligações químicas por reacções de condensação, consumindo energia sob a forma de ATP. |
A partir destas categorias principais, as enzimas ainda são subdivididas em outras categorias, podendo ser identificadas pelo seu número da EC; por exemplo, EC 5.4.2.2 é a fosfoglicomutase.
Aplicações |
Enzimas digestivas tais como a amilase, protease e lipase, reduzem os alimentos em componentes menores que são mais facilmente absorvidos no tracto digestivo.
As enzimas contribuem enormemente para inúmeras indústrias. Enzimas de processamento alimentar tais como a glucoamilase podem reduzir o alimento em glicose.
Uma aplicação industrial é a produção de antibióticos em larga escala. Encontram-se também determinados tipos de enzimas em produtos de limpeza, para ajudar a digerir gorduras e proteínas presentes em nódoas.
Também são usadas em investigação laboratorial e na medição de concentrações de substâncias com interesse clínico (Patologia Clínica, análises clínicas).
Referências |
↑ Howard Hugues Media Institute, The Structural Biology Program, "RNA, The Enzyme"
↑ A. Radzicka, R. Wolfenden (1995), "A proficient enzyme.", Science, 6(267), p. 90-931
↑ René Antoine Ferchault de Réaumur (1752), "Observations sur la digestion des oiseaux", Histoire de l'academie royale des sciences, 1752p. 266, 461
↑ Williams, H. S. (1904) A History of Science: in Five Volumes. Volume IV: Modern Development of the Chemical and Biological Sciences Harper and Brothers (New York)
↑ J. Dubos (1951), "Louis Pasteur: Free Lance of Science, Gollancz." in K.L. Manchester (1995), "Louis Pasteur (1822–1895)--chance and the prepared mind.", Trends in Biotechnology, 13(12), p.511-515
↑ Nobel Laureate Biography of Eduard Buchner at http://nobelprize.org Accessed 04 April 2007
↑ 1946 Nobel prize for Chemistry laureates at http://nobelprize.org Accessed 04 April 2007
↑ Blake CC, Koenig DF, Mair GA, North AC, Phillips DC, Sarma VR. (1965). «Structure of hen egg-white lysozyme. A three-dimensional Fourier synthesis at 2 Angstrom resolution.». Nature. 22 (206): 757-761. PMID 5891407 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Chen LH, Kenyon GL, Curtin F, Harayama S, Bembenek ME, Hajipour G, Whitman CP (1992). «4-Oxalocrotonate tautomerase, an enzyme composed of 62 amino acid residues per monomer». J. Biol. Chem. 267 (25): 17716-21. PMID 1339435 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Smith S (1994). «The animal fatty acid synthase: one gene, one polypeptide, seven enzymes». FASEB J. 8 (15): 1248–59. PMID 8001737
↑ Anfinsen C.B. (1973). «Principles that Govern the Folding of Protein Chains». Science: 223–230. PMID 4124164
↑ The Catalytic Site Atlas at The European Bioinformatics Institute Accessed 04 April 2007
↑ abc Voet, D., Voet, J.G. Bioquímica. 4º Edição. Porto Alegre: Artmed, 2013. 1482p.
↑ abc Lehninger Principles of Biochemistry (4th Ed.) Nelson, D., and Cox, M.; W.H. Freeman and Company, New York, 2005, ISBN 0-7167-4339-6
↑ Jaeger KE, Eggert T. (2004). «Enantioselective biocatalysis optimized by directed evolution.». Curr Opin Biotechnol. 15(4): 305–313. PMID 15358000
↑ Shevelev IV, Hubscher U. (2002). «The 3' 5' exonucleases.». Nat Rev Mol Cell Biol. 3 (5): 364–376. PMID 11988770
↑ Berg J., Tymoczko J. and Stryer L. (2002) Biochemistry. W. H. Freeman and Company ISBN 0-7167-4955-6
↑ Zenkin N, Yuzenkova Y, Severinov K. (2006). «Transcript-assisted transcriptional proofreading.». Science. 313: 518–520. PMID 16873663 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Ibba M, Soll D. (2000). «Aminoacyl-tRNA synthesis.». Annu Rev Biochem. 69: 617–650. PMID 10966471
↑ Rodnina MV, Wintermeyer W. (2001). «Fidelity of aminoacyl-tRNA selection on the ribosome: kinetic and structural mechanisms.». Annu Rev Biochem. 70: 415–435. PMID 11395413
↑ Firn, Richard. «The Screening Hypothesis - a new explanation of secondary product diversity and function». Consultado em 11 de outubro de 2006
↑ Fischer E. (1894). «Einfluss der Configuration auf die Wirkung der Enzyme». Ber. Dt. Chem. Ges. 27: 2985–2993
↑ Koshland D. E. (1958). «Application of a Theory of Enzyme Specificity to Protein Synthesis». Proc. Natl. Acad. Sci. 44 (2): 98–104. PMID 16590179
↑ Vasella A, Davies GJ, Bohm M. (2002). «Glycosidase mechanisms.». Curr Opin Chem Biol. 6 (5): 619–629. PMID 12413546 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Boyer, Rodney. «6». Concepts in Biochemistry (em English) 2nd ed. New York, Chichester, Weinheim, Brisbane, Singapore, Toronto.: John Wiley & Sons, Inc. pp. 137–138. ISBN 0-470-00379-0|acessodata=requer|url=(ajuda) !CS1 manut: Língua não reconhecida (link)
↑ Fersht, A (1985) Enzyme Structure and Mechanism (2nd ed) p50–52 W H Freeman & co, New York ISBN 0-7167-1615-1
↑ ab LEHNINGER, A. L. Princípios de bioquímica. São Paulo: Savier, 1985
↑ Eisenmesser EZ, Bosco DA, Akke M, Kern D. Enzyme dynamics during catalysis. Science. 2002 February 22;295(5559):1520–3. PMID 11859194
↑ Agarwal PK. Role of protein dynamics in reaction rate enhancement by enzymes. J Am Chem Soc. 2005 November 2;127(43):15248-56. PMID 16248667
↑ Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005 November 3;438(7064):117-21. PMID 16267559
↑ Yang LW, Bahar I. (5). «Coupling between catalytic site and collective dynamics: A requirement for mechanochemical activity of enzymes.». Structure. 13: 893–904. PMID 15939021 Verifique data em:|data=(ajuda)
↑ Agarwal PK, Billeter SR, Rajagopalan PT, Benkovic SJ, Hammes-Schiffer S. (5). «Network of coupled promoting motions in enzyme catalysis.». Proc. Natl. Acad. Sci. U S A. 99: 2794–9. PMID 11867722 Verifique data em:|data=(ajuda) !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Agarwal PK, Geist A, Gorin A. Protein dynamics and enzymatic catalysis: investigating the peptidyl-prolyl cis-trans isomerization activity of cyclophilin A. Biochemistry. 2004 August 24;43(33):10605-18. PMID 15311922
↑ Tousignant A, Pelletier JN. (agosto de 2004). «Protein motions promote catalysis.». Chem Biol. 11 (8): 1037–42. PMID 15324804
↑ Princípios de bioquímica, lehninger, 4 edição, São Paulo - SP
↑ LEHNINGER, A. L. Princípios de bioquímica.São Paulo: Savier, 1985
↑ Fisher Z, Hernandez Prada JA, Tu C, Duda D, Yoshioka C, An H, Govindasamy L, Silverman DN and McKenna R. (2005). «Structural and kinetic characterization of active-site histidine as a proton shuttle in catalysis by human carbonic anhydrase II.». Biochemistry. 44(4): 1097-115. PMID 15667203 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ AF Wagner, KA Folkers (1975) Vitamins and coenzymes. Interscience Publishers New York| ISBN 0-88275-258-8
↑ BRENDA The Comprehensive Enzyme Information System Accessed 04 April 2007
↑ Henri,V. (1902). «Theorie generale de l'action de quelques diastases». Compt. rend. hebd. Acad. Sci. Paris. 135: 916-919
↑ Sørensen,P.L. (1909). «Enzymstudien {II}. Über die Messung und Bedeutung der Wasserstoffionenkonzentration bei enzymatischen Prozessen». Biochem. Z. 21: 131-304
↑ Michaelis L., Menten M. (1913). «Die Kinetik der Invertinwirkung». Biochem. Z. 49: 333–369 English translation Visitado a 6 de Abril de 2007
↑ Briggs G. E., Haldane J. B. S. (1925). «A note on the kinetics of enzyme action». Biochem. J. 19: 339–339. PMID 16743508
↑ Radzicka A, Wolfenden R. (1995). «A proficient enzyme.». Science. 6 (267): 90–931. PMID 7809611
↑ Ellis RJ (2001). «Macromolecular crowding: obvious but underappreciated». Trends Biochem. Sci. 26 (10): 597-604. PMID 11590012
↑ Kopelman R (1988). «Fractal Reaction Kinetics». Science. 241 (4873): 1620–26. doi:10.1126/science.241.4873.1620
↑ Savageau MA (1995). «Michaelis-Menten mechanism reconsidered: implications of fractal kinetics». J. Theor. Biol. 176 (1): 115-24. PMID 7475096
↑ Schnell S, Turner TE (2004). «Reaction kinetics in intracellular environments with macromolecular crowding: simulations and rate laws». Prog. Biophys. Mol. Biol. 85 (2–3): 235-60. PMID 15142746
↑ Xu F, Ding H (2007). «A new kinetic model for heterogeneous (or spatially confined) enzymatic catalysis: Contributions from the fractal and jamming (overcrowding) effects». Appl. Catal. A: Gen. 317 (1): 70–81. doi:10.1016/j.apcata.2006.10.014
↑ Garcia-Viloca M., Gao J., Karplus M., Truhlar D. G. (2004). «How enzymes work: analysis by modern rate theory and computer simulations.». Science. 303 (5655): 186–195. PMID 14716003 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Olsson M. H., Siegbahn P. E., Warshel A. (2004). «Simulations of the large kinetic isotope effect and the temperature dependence of the hydrogen atom transfer in lipoxygenase». J. Am. Chem. Soc. 126 (9): 2820-1828. PMID 14995199 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Masgrau L., Roujeinikova A., Johannissen L. O., Hothi P., Basran J., Ranaghan K. E., Mulholland A. J., Sutcliffe M. J., Scrutton N. S., Leys D. (2006). «Atomic Description of an Enzyme Reaction Dominated by Proton Tunneling». Science. 312 (5771): 237–241. PMID 16614214 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Poulin R, Lu L, Ackermann B, Bey P, Pegg AE. Mechanism of the irreversible inactivation of mouse ornithine decarboxylase by alpha-difluoromethylornithine. Characterization of sequences at the inhibitor and coenzyme binding sites. J Biol Chem. 1992 Jan 5;267(1):150–8. PMID 1730582
↑ Ball, Philip (2006) The Devil's Doctor: Paracelsus and the World of Renaissance Magic and Science. Farrar, Straus and Giroux ISBN 0-374-22979-1
↑ Yoshikawa S and Caughey WS. (maio de 1990). «Infrared evidence of cyanide binding to iron and copper sites in bovine heart cytochrome c oxidase. Implications regarding oxygen reduction.». J Biol Chem. 265 (14): 7945–7958. PMID 2159465
↑ Hunter T. (1995). «Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling.». Cell. 80(2): 225–236. PMID 7834742
↑ Berg JS, Powell BC, Cheney RE. (2001). «A millennial myosin census.». Mol Biol Cell. 12(4): 780–794. PMID 11294886 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Meighen EA. (1991). «Molecular biology of bacterial bioluminescence.». Microbiol Rev. 55(1): 123–142. PMID 2030669
↑ Faergeman N. J, Knudsen J. (abril de 1997). «Role of long-chain fatty acyl-CoA esters in the regulation of metabolism and in cell signalling». Biochem J. 323: 1–12. PMID 9173866
↑ Doble B. W., Woodgett J. R. (abril de 2003). «GSK-3: tricks of the trade for a multi-tasking kinase». J. Cell. Sci. 116: 1175–1186. PMID 12615961
↑ Carr C. M., Kim P. S. (abril de 2003). «A spring-loaded mechanism for the conformational change of influenza hemagglutinin». Cell. 73: 823–832. PMID 8500173
↑ Phenylketonuria: NCBI Genes and Disease Accessed 04 April 2007
Ver também |
A Wikipédia tem o portal:
|
- Biossíntese
- Enovelamento de proteínas
- Desnaturação
- Química de Proteínas
- Metaloproteína
- Abzima


